Клиническая картина
Клиническая картина
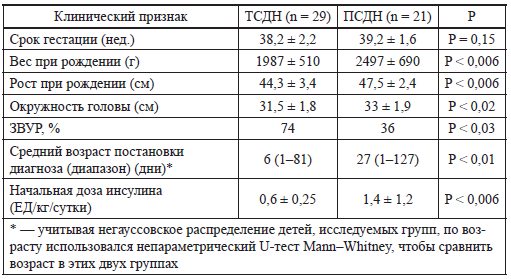
Для неонатального диабета характерна стойкая гипергликемия, отсутствие или плохие прибавки массы тела, у некоторых детей отмечается выраженное обезвоживание, кетоацидоз с выраженностью до комы. Характерны низкие концентрации инсулина в крови. При транзиторном сахарном диабете, в отличие от перманентного, дети чаще рождаются с задержкой физического развития (табл. 19). Задержка физического развития у плодов особенно выражена в последний триместр беременности. Интересно, что у мальчиков ЗВУР выражена более значительно, чем у девочек [74].
При дополнительном лабораторном обследовании у детей с транзиторным диабетом не обнаруживают антител к ?-клеткам поджелудочной железы и гаплотипам 2-го класса HLA, ассоциированных с диабетом 1-го типа. Экзокринная недостаточность поджелудочной железы нарушена не у всех детей. Однако клеточный дефект при ТСДН в настоящее время не установлен. Большинство больных поправляются в течение года, но некоторые имеют персистирующие нарушения толерантности к глюкозе или развивают в более старшем возрасте сахарный диабет. Не совсем ясен у данной категории больных патогенез диабета: дефицит инсулина и/или резистентность к нему. В более старшем возрасте постоянная гипергликемия, требующая терапии инсулином, развивается у большинства больных. Так, Metz С. et al. (2002) [149], показали, что сахарный диабет развился у 5 из 7 больных, перенесших ТСДН, в возрасте старше 8 лет. В 2000 году Temple I. К. et al. [196] продемонстрировали, что сахарный диабет развился у 11 из 18 больных, имевших транзиторный неонатальный диабет, после 4 лет жизни. Анализируя представленные в указанных работах данные, Polak М., Cave Н. (2007) [175] приходят к заключению, что транзиторный неонатальный диабет является постоянным дефектом клеток поджелудочной железы с переменной экспрессией в течении роста и развития.
Таблица 19 Клинические особенности притранзиторном (ТСДН) и перманентном (ПСДН) сахарном диабете новорожденных (BrunkowM. Е. et al., 2001) [47]
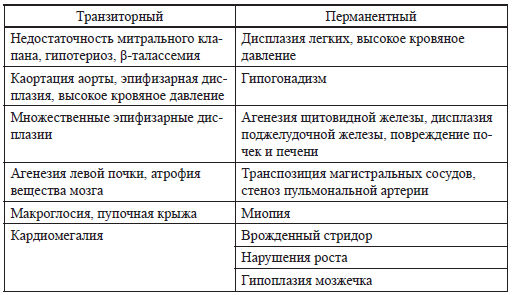
Хотя при обеих формах неонатального диабета достаточно часто встречаются пороки развития, но фенотипически дети отличаются (табл. 20).
Что касается, перманентного сахарного диабета, то в исследовании, проведенном IafuscoD. et al. (2002) [112] было продемонстрировано, что дети, заболевшие в первое полугодие жизни, в 6 раз чаще имели «защитные» аллели HLA-системы против классического диабета 1-го типа, по сравнению с детьми, заболевшими во втором полугодии. Кроме того, у детей, заболевших рано, в 5 раз реже встречались аутоиммунные маркеры.
Транзиторный сахарный диабет обычно развивается спорадически, хотя треть больных имеют установленную генетическую предрасположенность по линии отца. При этом не все отцы больны диабетом (von Muhlendahl К. Е., Herkenhoff Н., 1995) [155]. Некоторые больные имеют частичное удвоение длинного плеча 6-й отцовской хромосомы (6q24). В этой области находятся два гена (Arima Т. et al., 2001) [29]. Один кодирующий фактор транскрипции ZAC , регулирующий деление клетки и апоптоз, а также гипофизарную аденилатциклазу, активирующую полипептидный рецептор 1 и являющуюся мощным активатором секреции инсулина. Второй ген — HYMAI , функция которого в настоящее время неизвестна. В 2002 году было произведено экспериментальное моделирование ТСДН (Ма D. et al., 2002). В геном мыши был внедрен локус 6q24. У мышат развилось состояние, клинически очень сходное с транзиторным сахарным диабетом новорожденных, хотя и имевшее особенности.
Таблица 20
Фенотипические особенности детей при двух формах неонатального диабета
(Marquis Е. et al., 2002) [141]
Отцовская передача данного локуса приводила к развитию гипергликемии и увеличению риска развития диабета у мышат. Интересно, что данный эксперимент установил уменьшение экспрессии ключевого фактора транскрипции PDX-1 в эмбриональной поджелудочной железе мышей. Однако конкретный клеточный дефект; приводящий к нарушению синтеза инсулина, остается неизвестным.
При перманентном сахарном диабете, по крайней мере, у 50 % больных установлена мутация в гене, кодирующем субъединицу АТФ-чувствительных калиевых каналов, находящихся в ?-клетках поджелудочной железы. Наличие этой мутации приводит к тому что канал находится постоянно в открытом состоянии, что делает невозможным выделение инсулина (рис. 13–14). В настоящее время описано 9 мутаций в указанном гене. Некоторые из этих мутаций встречаются и при транзиторном сахарном диабете и диабете 2-го типа у более старших детей, что заставляет предполагать их генетическую общность (Gloyn A. L. et al., 2005 [86]; Babenko А. Р et al., 2006 [32]).
В настоящее время выделяют многочисленные наследственные синдромы, ассоциированные с перманентным сахарным диабетом новорожденных.
Агенезия поджелудочной железы и промоутер-фактор 1 инсулина (IPF1). Впервые ребенок с установленным нарушением гена, кодирующего промоутер-фактор 1 инсулина, и агенезией поджелудочной железы был описан Jonsson J. et al. в 1994 году [115]. Ребенок был гомозиготен по делеции единственного нуклеотида в 63 кодоне. Данный фактор регулирует эндокринное и экзокринное развитие поджелудочной железы. В 2000 году Vaulont S. et al. [203] показали, что он является регулятором инсулина и экспрессии гена соматостатина.
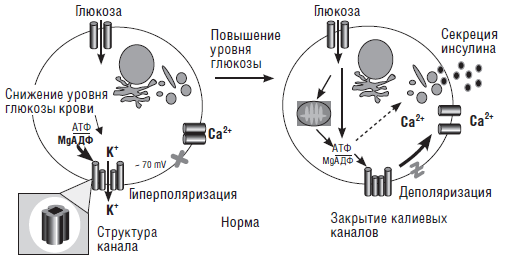
Рис. 13. Механизм секреции инсулина у здоровых людей (Polak М., Cave Н., 2007)
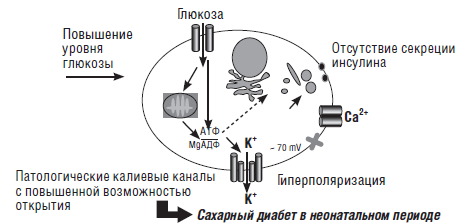
Рис. 14. Изменение секреции инсулина при генетически измененном калиевом канале (Polak М., Cave Н., 2007) [175]
Ранее Stoffers D. A. et al. (1998) [192] была описана семья, имевшая в 6 поколениях 8 больных с ранним началом диабета 2-го типа. Они были идентифицированы как гетерозиготы по той же мутации. Исследования, проведенные в последнее десятилетие во Франции, показали, что 6 % больных диабетом 2-го типа имеют мутации в гене, кодирующем промоутер-фактор 1 инсулина.
Гомозиготные аномалии в гене, кодирующем глюкокиназу. Мутации в указанном гене обычно приводят к умерено выраженной гипергликемии (Velho G., Froguel P., 1998) [204]. Глюкокиназа является регулятором метаболизма в ?-клетках поджелудочной железы, контролирует уровень секреции инсулина. Njolstad P. R. et al. [161] в 2001 году описали две семьи (норвежскую и итальянскую), имевших больных различными типами диабета. В этих семьях родились дети, у которых в первый день жизни манифестировал классический перманентный сахарный диабет. При анализе оказалось, что они являются гомозиготами по мутации в гене, кодирующем глюкокиназу. Их родители были гетерозиготами по той же мутации и обладали пониженной толерантностью к глюкозе. Polak М., Cave Н. (2007) [175] считают, что если оба родителя имеют пониженную толерантность к глюкозе, то они должны быть обследованы на наличие мутаций в указанном гене.
Иммунная днзрегуляцня, полиэндокринопатия, энтеропатия, Х-связанная — IPEX-синдром. В 1996 году Peake J. Е. et al. [171] сообщили о мутации X хромосомы, сопровождающейся эксфолиативным дерматитом, диареей, гемолитической анемией, аутоиммунным поражением щитовидной железы и неонатальным диабетом. Дети умирают на первом году жизни от сепсиса. У некоторых детей описана агенезия островков Лангерганса. Аутоиммунная природа этого синдрома подтверждалась успешной терапией цитостатиками (Satake N. et al., 1993 [182]; Baud О. et al., 2001 [35]). Позже у больных были идентифицированы антитела к глутаминовой декарбоксилазе. Применение иммуносупрессивной терапии как этапа подготовки к трансплантации костного мозга приводило к купированию сахарного диабета, а затем диареи и дерматита. Установлено, что данная мутация, по крайней мере, в эксперименте, приводит к усиленной пролиферации СБ4+/СБ8-Т-лимфоцитов с мультиорганной инфильтрацией. В результате этого развивается гемофагоцитический синдром, и мальчики без лечения погибают на 15-25-й день жизни после рождения (Brunkow М. Е. et al., 2001) [47]. Описано три случая выздоровления после трансплантации красного костного мозга при данной патологии [35].
Синдром Wolcott-Rallison. Синдром назван в честь врачей Wolcott С. D., Rallison М. V. впервые в 1972 году, описавших трех больных в одной семье, имевших сочетание неонатального диабета и спондилоэпифизарных нарушений скелета [207]. Это аутосомно-рецессивное заболевание, характеризующееся ранним началом сахарного диабета, обычно в неонатальный период, и спондилоэпифизарными нарушениями роста. У больных часто отмечается гепатомегалия, задержка умственного развития, почечная недостаточность, ранняя смерть. В 2000 году Delepine М. et al. [66] описали двух подобных детей в разных семьях, имеющих мутацию 2р12. В данном локусе находится ген, являющийся регулятором синтеза инсулина и белков в ?-клетках поджелудочной железы. В мире к 2010 году были описаны около 60 пациентов с синдромом Wolcott-Rallison (Julier С., Nicolino М., 2010) [116]. Хотя, конечно, больных небольшое количество, но, тем не менее, некоторые закономерности течения данного синдрома понятны. Сахарный диабет развивается в первые шесть месяцев жизни, спондилоэпифизарные нарушения роста начинают проявляться в первые два года жизни. Они выражены как клинически, так и рентгенологически (рис. 15–16).
Другие проявления более вариабельны, но среди них можно выделить: острую печеночную недостаточность, почечные дисфункции, экзокринную недостаточность поджелудочной железы, задержку ПМР, гипотиреоз, нейтропению, рецидивирующие инфекции. Достаточно часто отмечаются переломы костей.
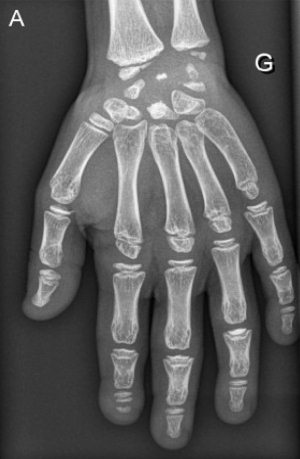
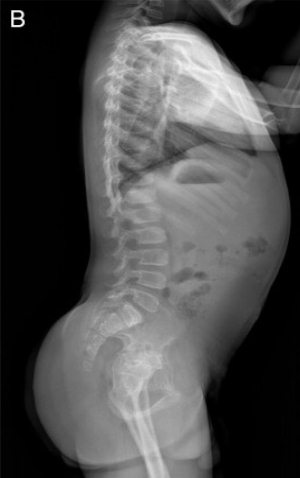
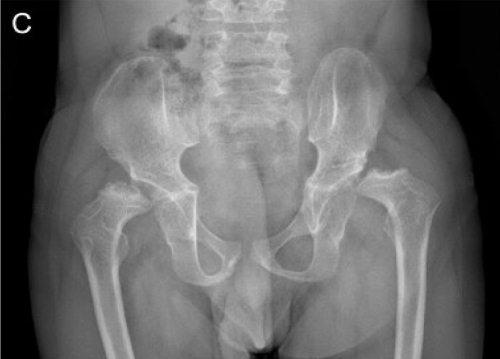
Рис. 15. Рентгенограммы пациента с синдромом Wolcott-Rallison (возраст 13 лет)
(Julier С., Nicolino М., 2010) [116].
А — руки: кости запястья небольшие с дисплазией дистального отдела лучевого и локтевого эпифизов; несколько фаланг являются диспластическими с аномальными укорочениями метафизов в проксимальном отделе. Б — позвоночник: в грудном отделе позвоночника имеются уплощения тел позвонков с дефектами переднего края. В — таз: гипоплазия вертлужной впадины с дисплазией эпифизов бедренных костей
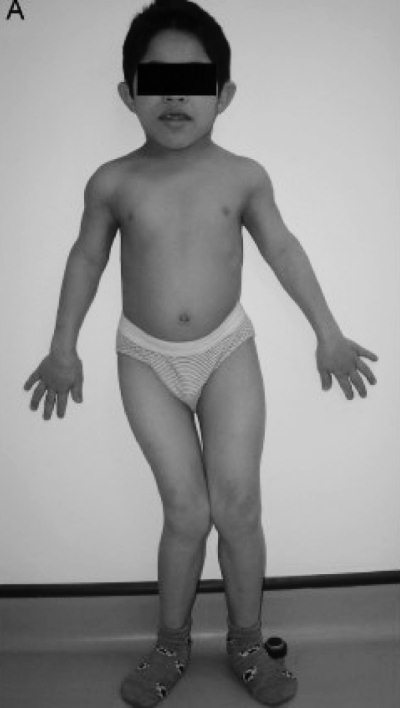
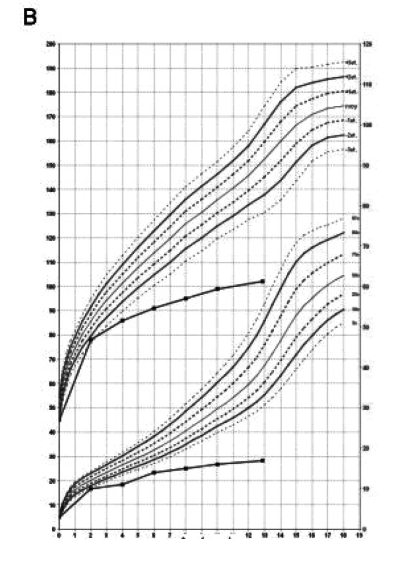
Рис. 16. Слева скелетные дисплазии и спондилоэпифизарные нарушения роста у пациента с синдромом Wolcott-Rallison (возраст 13 лет). Справа — диаграмма, демонстрирующая нарушения роста (Julier С., Nicolino М., 2010) [116]
Большая часть больных, рожденных от близкородственных браков, описана на Ближнем Востоке, Северной Африке, Пакистане и Турции. В настоящее время считают, что данный синдром должен быть исключен у любого ребенка, с диагностированным неонатальным сахарным диабетом (Rubio-Cabezas О. et al., 2009) [181]. Возможна генетическая и пренатальная диагностика данного синдрома (Stoy J. et al., 2007) [193]. Прогноз для жизни у больных неблагоприятный: описан только один больной, доживший до 35 лет (Senee V. et al, 2004 [185]; Ozbek М. N. et al, 2010 [168]). Christen H. J. et al. [52] в 1992 году описали двух мальчиков с Х-связанной высокой активностью фосфорибозил-АТФ-пирофосфатазой, развивших перманентный сахарный диабет в первый день жизни. Нарушенная толерантность к глюкозе сохранилась в течение жизни, хотя бывали периоды ремиссии, и больные не нуждались в инсулине. Оба ребенка кроме сахарного диабета имели задержку психического развития, атаксию и прогрессивную аксональную невропатию. Матери мальчиков страдали подагрой и гестационным диабетом.
Yorifuji Т. et al. [209] в 1994 году опубликовали клиническое описание новорожденных детей в японской семье с перманентным сахарным диабетом ассоциированным с выраженной гипоплазией поджелудочной железы (головки и хвоста), врожденным «синим» пороком сердца. Не все дети в описанной семье имели манифестирование сахарного диабета в неонатальном периоде. Клинические проявления и манифест диабета зависели от степени гипоплазии поджелудочной железы. Авторы считают, что ими был описан новый наследственный синдром. Больше описаний подобных случаев мы в литературе не нашли.
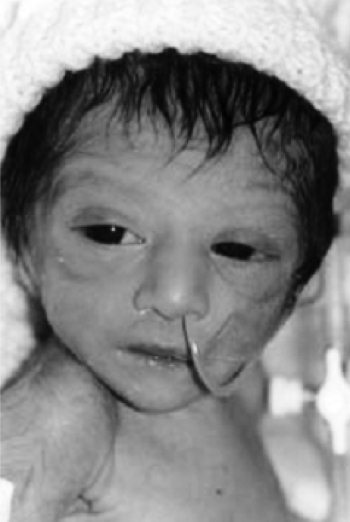
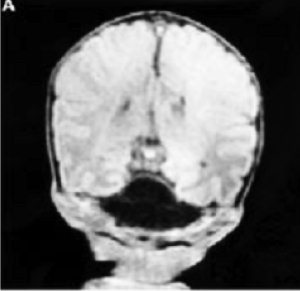
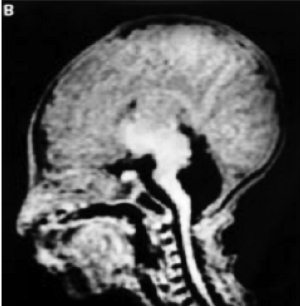
Рис. 17. Фотография и данные ЯМР ребенка с перманентным сахарным диабетом новорожденных и гипоплазией мозжечка (Hoveyda N. et al., 1999) [109]
В 1999 году была описана семья, имевшая 3 детей с перманентным сахарным диабетом новорожденных и гипоплазией мозжечка (Hoveyda N. et al., 1999) [109] (рис. 17). Установлен аутосомно-рецессивный тип наследования. Все дети не дожили до 1 года и погибли от метаболических расстройств, дыхательной недостаточности, сепсиса. Позже Sellick G. S. et al. (2004) [183] было установлено, что есть много специфических активаторов транскрипции, регулирующих экспрессию генов в ?-клетках поджелудочной железы и нейронов.
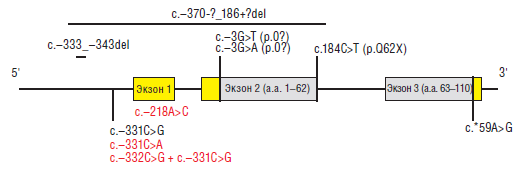
Рис. 18. Схема гена человека, кодирующего инсулин (Garin I. et al., 2010) [81]. Схема человеческого гена, кодирующего инсулин с указанием расположения мутаций при гомозиготном или гетерозиготном наследовании у пробандов с сахарным диабетом. Мутации с участием кодирующих областей приведены выше гена, с участием некодирующих областей ниже гена. Белки в кодирующих областях гена, обозначены серым цветом, а регионы кодирования 5'— и З'-нетранслируемой области мРНК инсулина — желтым цветом. Красным и черным цветом обозначены мутации, обнаруженные у пробандов с транзиторным и перманентным сахарным диабетом новорожденных, соответственно. Экзон 2 кодирует 1-62 аминокислоты, а 3 экзон — 63-110 аминокислоты в молекуле препроинсулина
Данный синдром связан с мутацией PTF1A фактора транскрипции, регулирующего развитие поджелудочной железы, а также экспрессирующегося в мозжечке. Недавно, в 2006 году, мутации, описанные Senee V. et al. [184], в другом гене транскрипции (Glis3 ) объяснили синдром, имеющий комплекс признаков: сахарный диабет новорожденных, гипотиреоз, врожденную глаукому, кисты в почках, фиброз печени.
Garin I. et al. [81] в 2010 году сообщили о рецессивных мутациях в гене, кодирующем инсулин, что приводило к развитию неонатального сахарного диабета. Эти мутации влияют на биосинтез проинсулина и функционируют как нулевые мутации. Одна мутация приводит к удалению из гена инсулина экзонов 1 и 2. Есть несколько мутаций, расположенных в промоторной области гена, которые приводят к снижению транскрипции. Мутации в области кодирования З’-нетранслируемой области мРНК влияют на стабильность мРНК. Другие мутации затрагивают инициацию трансляции мРНК инсулина или приводят к синтезу усеченной молекулы проинсулина (рис. 18).
В 2010 году Nicolino М. et al. [160] провели генетическое исследование семьи, имевшей трех детей, больных неонатальным сахарным диабетом. В результате были выявлены новые мутации в гомеобоксных генах поджелудочной железы (PDX1, IPF1) (рис. 19).
Имеются единичные описания (Otonkoski Т. et al., 2000) [166], указывающие, что инфицирование энтеровирусом (ECHO вирус 6) беременной женщины в конце первого триместра беременности приводит к развитию аутоиммунных реакций с развитием сахарного диабета у новорожденного и присутствием антиинсулиновых антител, а также антител к карбоксилазе глутаминовой кислоты при рождении или сразу после рождения. У больной девочки, описанной в указанном исследовании, имелась гипоплазия поджелудочной железы, возможно, вызванная действием вируса или указанных антител.
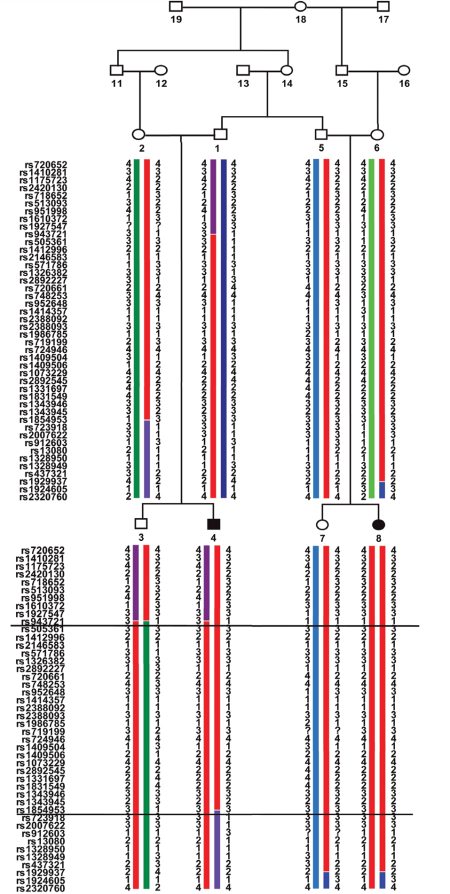
Рис. 19. Родословная и схема генов у больных с неонатальным сахарным диабетом
(NicolinoM.etal.,2010) [160]
Анализ родословной и генетический анализ у больных неонатальным диабетом (больные дети обозначены черным цветом). Регион, размером 4,4 Мб имеет мутации, обозначенные красным цветом (гомозиготные гаплотипы у больных)
Наконец, перманентный неонатальный сахарный диабет может входить в структуру митохондриальных болезней (Metz С. et. al, 2002) [149]. В 2011 году польские ученые Pronicka Е. et al. [176] описали новые мутации, приводящие к дефициту деоксигуанозин-киназы, являющиеся причиной гепа-тоцеребральной формы митохондриального синдрома. Дети родились со ЗВУР, у всех развилась печеночная недостаточность, явившаяся непосредственной причиной смерти. В неонатальный период дети имели неврологическую симптоматику (мышечную гипотонию, задержку ПМР, птоз). Среди лабораторных показателей обращали на себя внимание гипогликемия и метаболический ацидоз. Повышение уровня трансферрина, ферритина и альфа-фетопротеина напоминало изменения, характерные для гемохроматоза новорожденных. При гистологическом исследовании печени выявлен цирроз печени с выраженным фиброзом и полной потерей архитектуры печени. В поджелудочной железе обнаружены участки гиперплазии клеток.
Интересное наблюдение приводят Abaci A. et al. (2010) [23]. Ребенок поступил на отделение интенсивной терапии в возрасте одного месяца с жалобами на лихорадку, вялость, плохой аппетит. Он родился в срок, с весом 2550 г. Сразу же после родов приложен к груди и находился на грудном вскармливании. На 4-е сутки жизни мальчик был выписан домой. При поступлении на отделение ребенок был раздражительным, сонливым. Выявлена обезвоженость (потеря веса 10 %), отмечена тахикардия (176 уд. в мин), тахипноэ (64 дых. в мин), температура тела 39,1 °C (ректально), АД = 65/45 мм рт. ст. С учетом признаков системного воспалительного ответа, ребенку был поставлен диагноз: «сепсис». Мы не будем полностью описывать обследование и полученные результаты анализов, но оказалось, что у ребенка глюкоза крови — 1125 мг/дл, концентрация инсулина — 9 jiU/мл, а С-пептид — 0,5 нг /мл. Также был выявлен кетоацидоз. С учетом клинической картины и лабораторных показателей, у младенца был заподозрен неонатальный сахарный диабет, начато введение инсулина в дозе 0,05 МЕ/кг/час в течение 2 часов, с переходом на 0,1 ЕД/кг/час. Гипергликемию и кетоацидоз купировали в течение 14 часов. Ребенок был выписан домой через три недели на поддерживающей дозе инсулина.
Все вышеперечисленные исследования подтверждают гипотезу, что неонатальный сахарный диабет — это большая группа гетерогенных заболеваний, часто имеющих генетическую природу. Необходимо отметить, что многие заболевания новорожденных являются гетерогенными: сепсис [21], БЛД [20], болезнь гиалиновых мембран [1], ретинопатия недоношенных [8], ДВС-синдром 22] и др.
Более 800 000 книг и аудиокниг! 📚
Получи 2 месяца Литрес Подписки в подарок и наслаждайся неограниченным чтением
ПОЛУЧИТЬ ПОДАРОКДанный текст является ознакомительным фрагментом.
Читайте также
КЛИНИЧЕСКАЯ КАРТИНА
КЛИНИЧЕСКАЯ КАРТИНА От проникновения возбудителя в организм до развития клинических проявлений инфекции проходит около 2 недель, но инкубационный период может удлиняться до 25 дней. В зависимости от локализации поражения встречаются разные клинические формы инфекции:
46. Клиническая картина глаукомы
46. Клиническая картина глаукомы Открытоугольная глаукома обычно возникает после сорока лет. Начало заболевания нередко бессимптомное. 15–20 % больных жалуются на появление радужных кругов вокруг источника света, периодическое затуманивание зрения. Нередко наблюдается
Клиническая картина болезни
Клиническая картина болезни Клиническая картина болезни занимает центральное место во всех учебных руководствах по клинической гомеопатии и на курсах специализации по гомеопатии для врачей или студентов. Переход от диагностики заболевания к гомеопатическому поиску
Клиническая картина приступа
Клиническая картина приступа Две хорошо различимые формы течения (с некоторыми переходами) указывают на соответствующее средство: сухая спастическая астма и влажная астма, астматический бронхит.Сухая спастическая астма протекает с продукцией небольшого количества
Клиническая картина
Клиническая картина Известно, что концентрация глюкозы у новорожденного в крови вены пуповины составляет от 60 до 80 % от концентрации в венозной крови матери. Сразу же после рождения ее концентрация снижается, а через 2–3 часа после рождения начинает повышаться и
Клиническая картина
Клиническая картина Для неонатального диабета характерна стойкая гипергликемия, отсутствие или плохие прибавки массы тела, у некоторых детей отмечается выраженное обезвоживание, кетоацидоз с выраженностью до комы. Характерны низкие концентрации инсулина в крови. При
Клиническая картина
Клиническая картина Отмечается в первую очередь повышенный тонус миометрия матки еще до начала родовой деятельности и во время родов. Поэтому затрудняется пальпация предлежащей части плода к входу в малый таз. Как правило, у таких родильниц происходит преждевременное
КЛИНИЧЕСКАЯ КАРТИНА
КЛИНИЧЕСКАЯ КАРТИНА Исторические замечания Уже в древней литературе можно найти меткие описания шизофрении. Например, в Священном Писании выделяются два основных симптома шизофрении — аутизм и расщепление: «…встретил Его вышедший из гробов человек, одержимый
Нетипичная клиническая картина
Нетипичная клиническая картина Несмотря на большое психопатологическое разнообразие, можно дать приблизительное описание клинической картины шизофрении. Случаются, однако, нетипичные варианты, которые вызывают классификационные трудности. Их можно разделить на пять
Клиническая картина
Клиническая картина Геморрой у женщин и у мужчин имеет постепенное развитие и хроническое течение, обостряющееся острыми приступами. Выделяют три группы геморроя: внутренний, наружный и комбинированный.Симптомами геморроя являются жжение и зуд в области заднего
Клиническая картина
Клиническая картина На ранних стадиях заболевания у детей отмечается полиурия, причем у маленьких детей она рассматривается как ночное недержание мочи. Встречается также и полидипсия: после того как белье высыхает, оно становится жестким, словно подкрахмаленным.
Клиническая картина
Клиническая картина Хронический гастрит очень часто возникает у больных с гастроэнтерологической патологией. В данном случае он будет выражен воспалением слизистой желудка; сопутствующие факторы – нарушение моторной, секреторной и некоторых других функций. Очень
Клиническая картина
Клиническая картина Для того чтобы правильно понять и оценить происходящие в организме патологические процессы, их принято правильно классифицировать. Поэтому приведем несколько слов о классификации межпозвоночных грыж.Если коротко, то в медицинской литературе
КЛИНИЧЕСКАЯ КАРТИНА
КЛИНИЧЕСКАЯ КАРТИНА Основной жалобой при наличии бронхоэктазов является постоянный кашель с большим количеством («полным ртом») гнойной мокроты, которую маленькие дети часто проглатывают. Дети старше 3 – 4 лет откашливают мокроту. Носовое дыхание затруднено; характерно
КЛИНИЧЕСКАЯ КАРТИНА
КЛИНИЧЕСКАЯ КАРТИНА Выделяют следующие формы муковисцидоза:• преимущественно легочная форма с минимальными проявлениями поражения кишечника (15 – 20%);• преимущественно кишечная форма (5 – 10%);• смешанная форма (75-80%);• мекониальный илеус (5-19%);• стертые формы
КЛИНИЧЕСКАЯ КАРТИНА
КЛИНИЧЕСКАЯ КАРТИНА Клинически астма проявляется приступообразным, навязчивым, часто ночным, кашлем, свистящим, затрудненным дыханием, одышкой и удушьем. Клиническая симптоматика при астме непостоянна и меняется в течение суток. Сухие, свистящие хрипы, а у детей раннего